from rdkit import Chem
from rdkit.Chem import rdChemReactions
from rdkit.Chem import Draw
from rdkit.Chem import rdDepictor
from rdkit.Chem.Draw import IPythonConsole
import rdkit
print(rdkit.__version__)2025.03.3We often get questions about how to relate atoms in the products of a chemical reaction to the corresponding atoms in the reactants which were provided to the reaction. The RDKit provides a couple of pieces of information that can help here; this post shows how to access that information and what it means.
from rdkit import Chem
from rdkit.Chem import rdChemReactions
from rdkit.Chem import Draw
from rdkit.Chem import rdDepictor
from rdkit.Chem.Draw import IPythonConsole
import rdkit
print(rdkit.__version__)2025.03.3This is the reaction definition Niementowski_quinazoline from the SI of the paper “A Collection of Robust Organic Synthesis Reactions for In Silico Molecule Design” by Hartenfeller et al. .
rxn = rdChemReactions.ReactionFromSmarts('[c:1](-[C;$(C-c1ccccc1):2](=[OD1:3])-[OH1]):[c:4](-[NH2:5]).[N;!H0;!$(N-N);!$(N-C=N);!$(N(-C=O)-C=O):6]-[C;H1,$(C-[#6]):7]=[OD1]>>[c:4]2:[c:1]-[C:2](=[O:3])-[N:6]-[C:7]=[N:5]-2')
rxn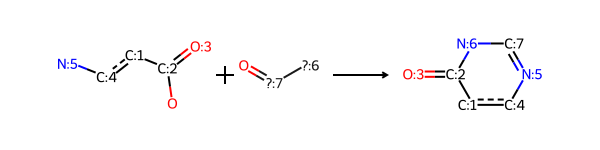
reactants = [Chem.MolFromSmiles(x) for x in ('c1c(C(=O)O)c(N)cc(CC)c1', 'C(=O)Nc1ccccc1')]
Draw.MolsToGridImage(reactants)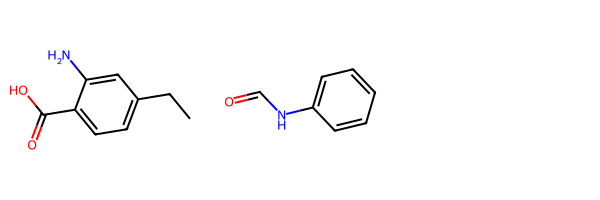
prods = rxn.RunReactants(reactants)
len(prods)1prods[0][0]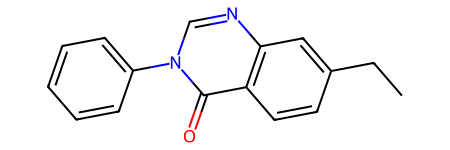
prods[0][0].GetAtomWithIdx(0).GetPropsAsDict(includeComputed=False){'old_mapno': 4, 'react_atom_idx': 5, 'react_idx': 0}old_mapno is the atom map number of the atom in the product template, react_idx is the index of the reactant that the atom came from (this was added in release 2024.09.6, so it won’t be there in older releases), and react_atom_idx is the index of the atom in that reactant.
Here’s an atom that wasn’t in the product template, so it doesn’t have an atom map number:
prods[0][0].GetAtomWithIdx(10).GetPropsAsDict(includeComputed=False){'react_atom_idx': 7, 'react_idx': 0}Here’s a sample function that draws a product molecule and highlights atoms and bonds based upon which reactant they came from. It can also optionally include the original atom indices.
from IPython.display import SVG
def draw_mol_highlighted_by_atom_origin(mol,
includeOriginalAtomIndices=True,
size=(450,450),
colors=[(1,1,0.67),(1,0.8,0.6),(1., .71, .76),(.8, 1., .8)],
):
# copy the molecule since we will modify it:
mol = Chem.Mol(mol)
if not mol.GetNumConformers():
rdDepictor.Compute2DCoords(mol)
rdDepictor.StraightenDepiction(mol)
atom_highlights = {}
bond_highlights = {}
rads = {}
mults = {}
for at in mol.GetAtoms():
pd = at.GetPropsAsDict()
if 'react_idx' in pd:
atom_highlights[at.GetIdx()] = [colors[pd['react_idx']]]
rads[at.GetIdx()] = 0.4
mults[at.GetIdx()] = 1
if includeOriginalAtomIndices and 'react_atom_idx' in pd:
at.SetProp('atomNote',str(pd['react_atom_idx']))
for bnd in mol.GetBonds():
bidx = bnd.GetBeginAtomIdx()
eidx = bnd.GetEndAtomIdx()
if bidx in atom_highlights and eidx in atom_highlights and atom_highlights[bidx] == atom_highlights[eidx]:
bond_highlights[bnd.GetIdx()] = atom_highlights[bidx]
d2d = Draw.MolDraw2DSVG(size[0],size[1])
d2d.DrawMoleculeWithHighlights(mol,'',atom_highlights,bond_highlights,rads,mults)
d2d.FinishDrawing()
return d2d.GetDrawingText()mol = prods[0][0]
SVG(draw_mol_highlighted_by_atom_origin(mol))
SVG(draw_mol_highlighted_by_atom_origin(mol,includeOriginalAtomIndices=False))