Doing extended Hueckel calculations with the RDKit
tutorial
Including an exploration of charge variability across conformers
Published
May 30, 2025
This is an updated and revised version of an older post.
The RDKit has included an optional integration with a semi-empirical quantum mechanics package called YAeHMOP since the 2019.03 release. If you build the RDKit yourself, this is disabled by default, but it is part of the RDKit conda-forge and pypi builds, so it’s available to most RDKit users.
There’s no documentation for this integration yet… thus this blog post.
Some background
One of my major projects in grad school was to write a software package for performing extended Hueckel calculations and analyzing and visualizing the results. I released the source for YAeHMOP (“Yet Another extended Hueckel Molecular Orbital Package”) way back when using some weird license (the open-source landscape in chemistry was “a bit” different back in the mid-90s). After changing fields, I more or less forgot about YAeHMOP until a couple of years ago when Patrick Avery contacted me about integrating it into Avogadro. To make things easier, I put the source up on Github and switched to a BSD license. Since that point we’ve done a bit of additional cleanup work on the eHT code (the visualization pieces are still there, but really only have historic interest) and made it useable as a library.
There are a couple of applications where I think it might be interesting to have access to quick, interpretable QM results from the RDKit. These include getting partial charges and starting to think about quick and dirty descriptors for things like bond strengths and reactivity. So I decided to do an integration of YAeHMOP into the RDKit.
There are a bunch of books and papers describing extended Hueckel theory and the various types of analysis that one can do with it; I’m not going to cite all of those here. There’s also no formal publication describing YAeHMOP (what a shock!), but there is a chapter in my PhD thesis that provides an fairly comprehensive tutorial overview of the theory.
from rdkit import Chemfrom rdkit.Chem import Drawfrom rdkit.Chem import rdDepictorfrom rdkit.Chem import rdDistGeomfrom rdkit.Chem import rdForceFieldHelpersfrom rdkit.Chem import rdMolAlignrdDepictor.SetPreferCoordGen(True)from rdkit.Chem.Draw import IPythonConsoleIPythonConsole.ipython_3d =True# this is the package including the connection to YAeHMOPfrom rdkit.Chem import rdEHTToolsimport numpy as npimport rdkitprint(rdkit.__version__)import timeprint(time.asctime())
2025.03.2
Tue May 27 06:32:31 2025
import matplotlib.pyplot as pltplt.rcParams['font.size'] ='16'%matplotlib inline
The reduced overlap population matrix provides the Mulliken overlap population between atoms in the molecule. This tells us something about the strength of the interaction between the atoms. It’s returned as a vector representing a symmetric matrix:
# convenience function to set a "MulilkenOverlapPopulation" property on bonds.def set_overlap_populations(m,ehtRes): rop = ehtRes.GetReducedOverlapPopulationMatrix()for bnd in m.GetBonds(): a1 = bnd.GetBeginAtom() a2 = bnd.GetEndAtom()if a1.GetAtomicNum()==1or a2.GetAtomicNum()==1:continue# symmetric matrix: i1 =max(a1.GetIdx(),a2.GetIdx()) i2 =min(a1.GetIdx(),a2.GetIdx()) idx = (i1*(i1+1))//2+ i2 bnd.SetDoubleProp("MullikenOverlapPopulation",rop[idx])
set_overlap_populations(mh,res)
Look at a few of the overlap populations:
for bnd inlist(mh.GetBonds())[:5]:ifnot bnd.HasProp("MullikenOverlapPopulation"):continueprint(f'{bnd.GetIdx()}{bnd.GetBeginAtom().GetSymbol()}({bnd.GetBeginAtomIdx()})-{bnd.GetEndAtom().GetSymbol()}({bnd.GetEndAtomIdx()}) {bnd.GetBondType()}{bnd.GetDoubleProp("MullikenOverlapPopulation") :.3f}')
0 C(0)-C(1) SINGLE 0.787
1 C(1)-C(2) AROMATIC 1.127
2 C(2)-C(3) AROMATIC 1.084
3 C(3)-C(4) AROMATIC 1.113
4 C(4)-N(5) SINGLE 0.720
The wrapper also currently provides the reduced charge matrix (described in that thesis chapter linked above). I’m not going to discuss that here.
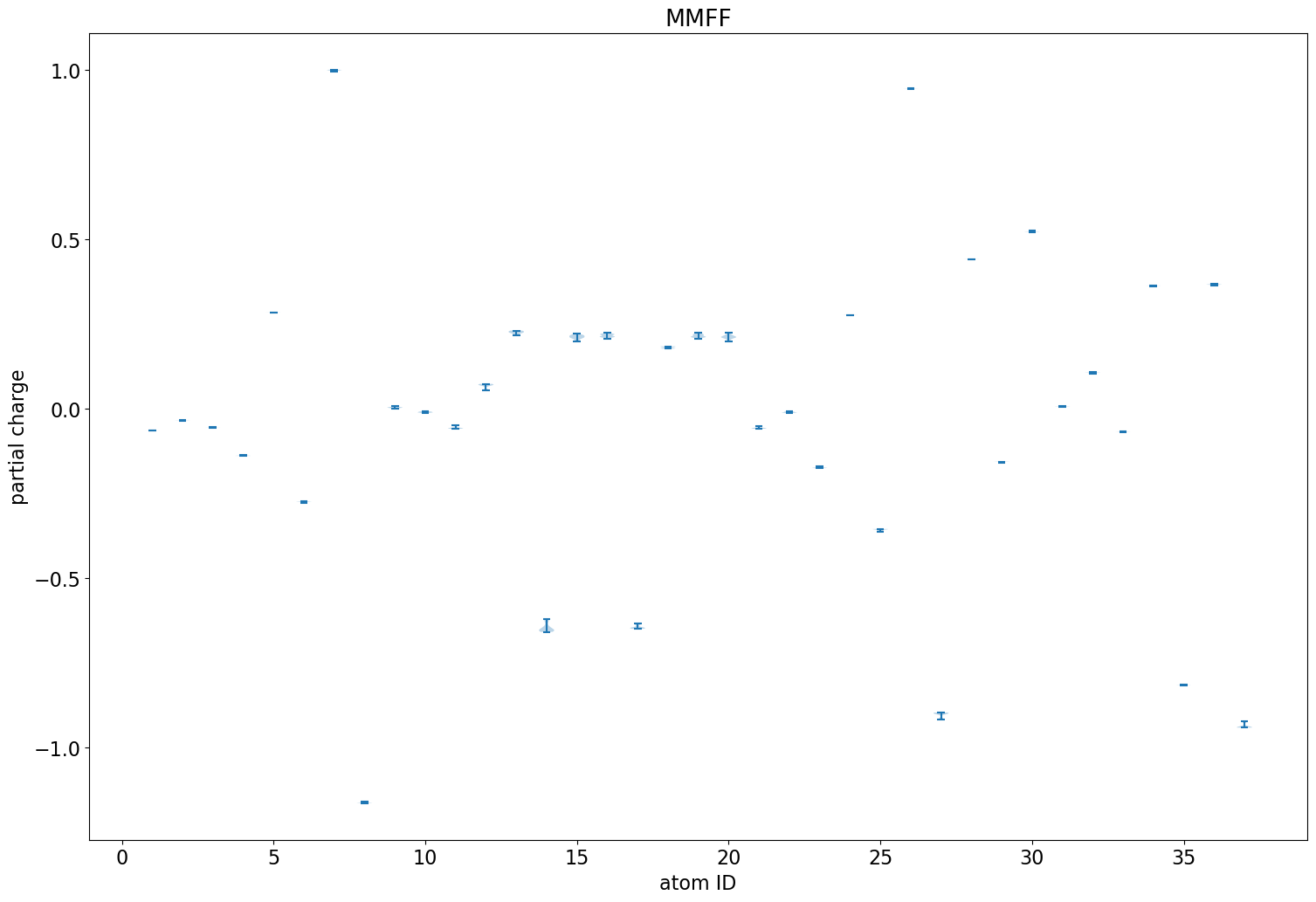
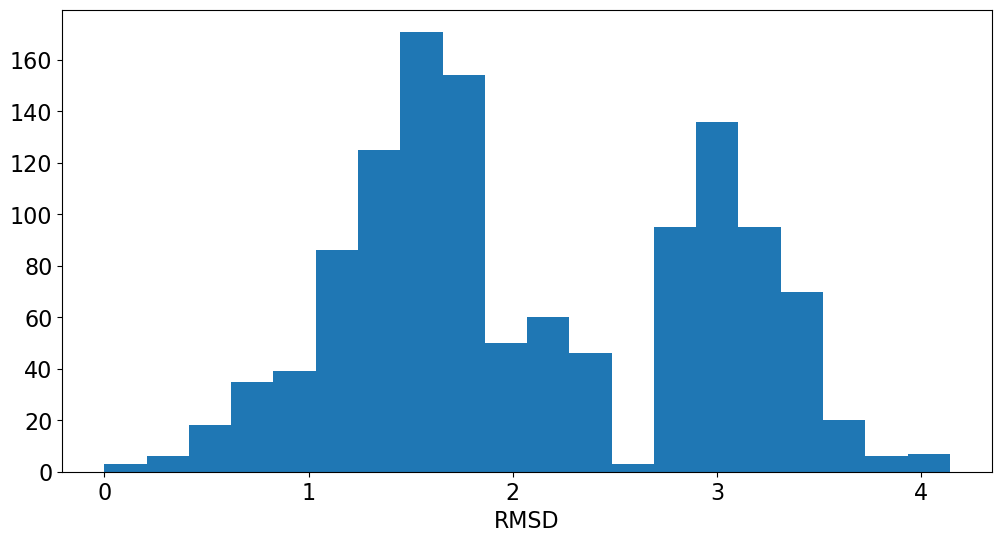
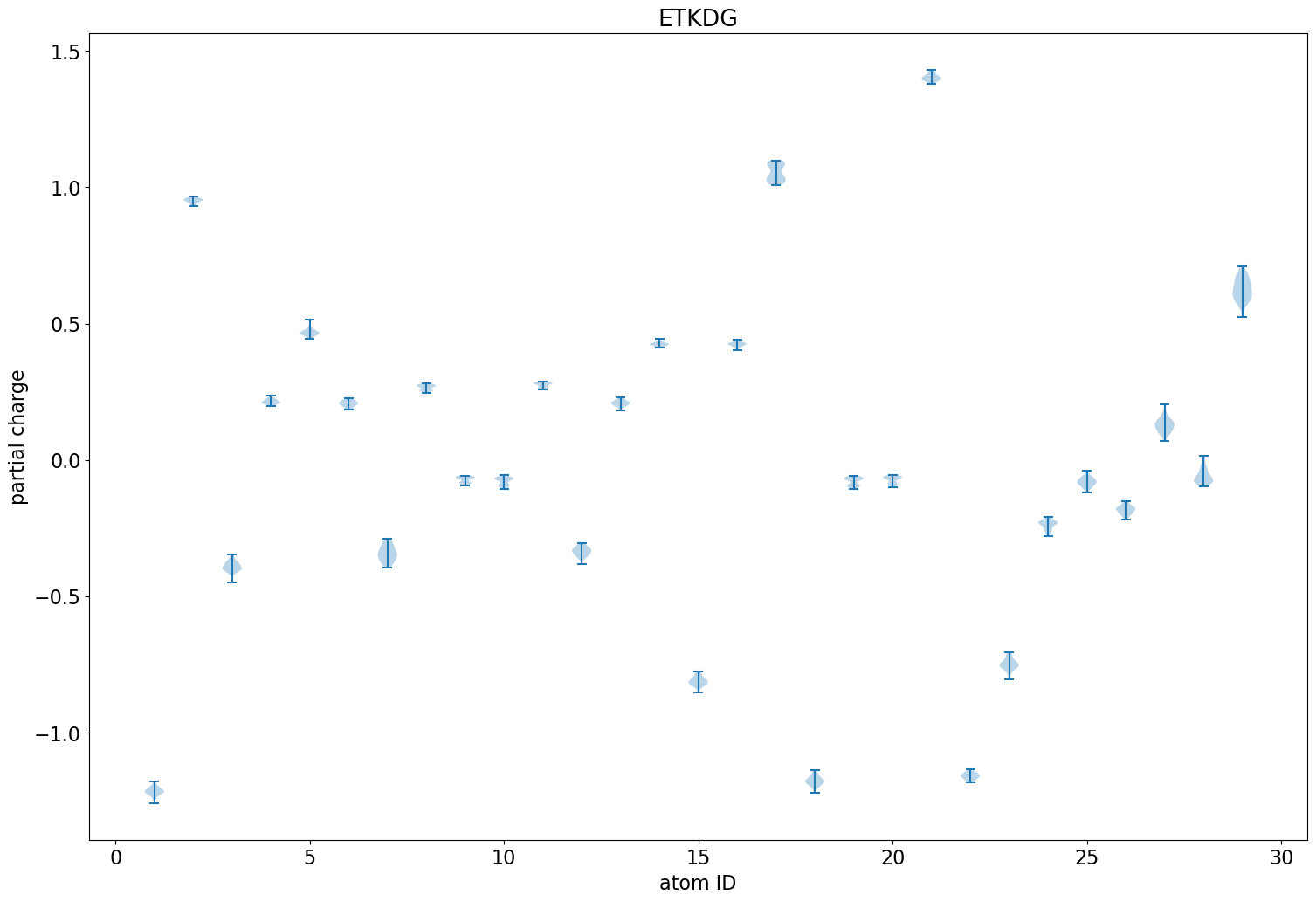
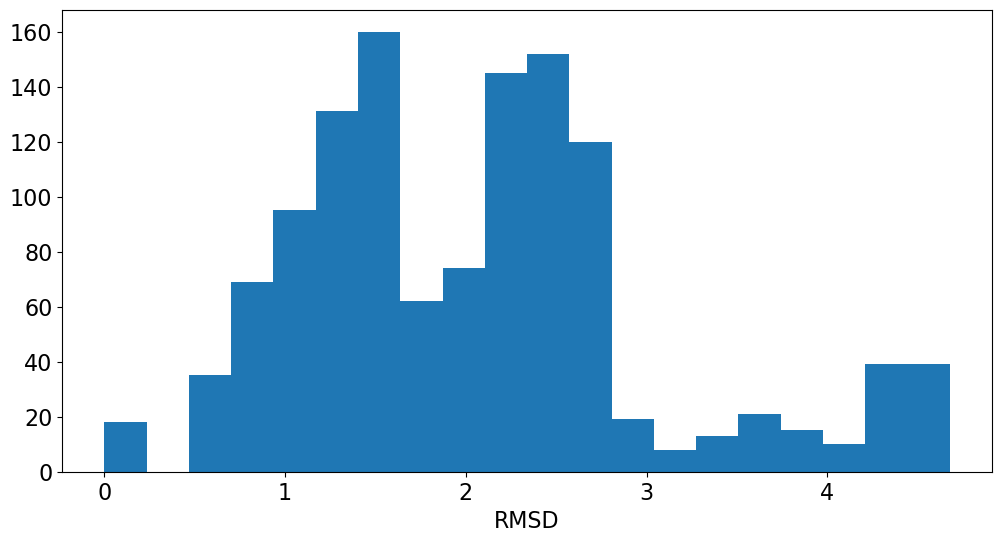
Application: Look at variability of charges between conformers
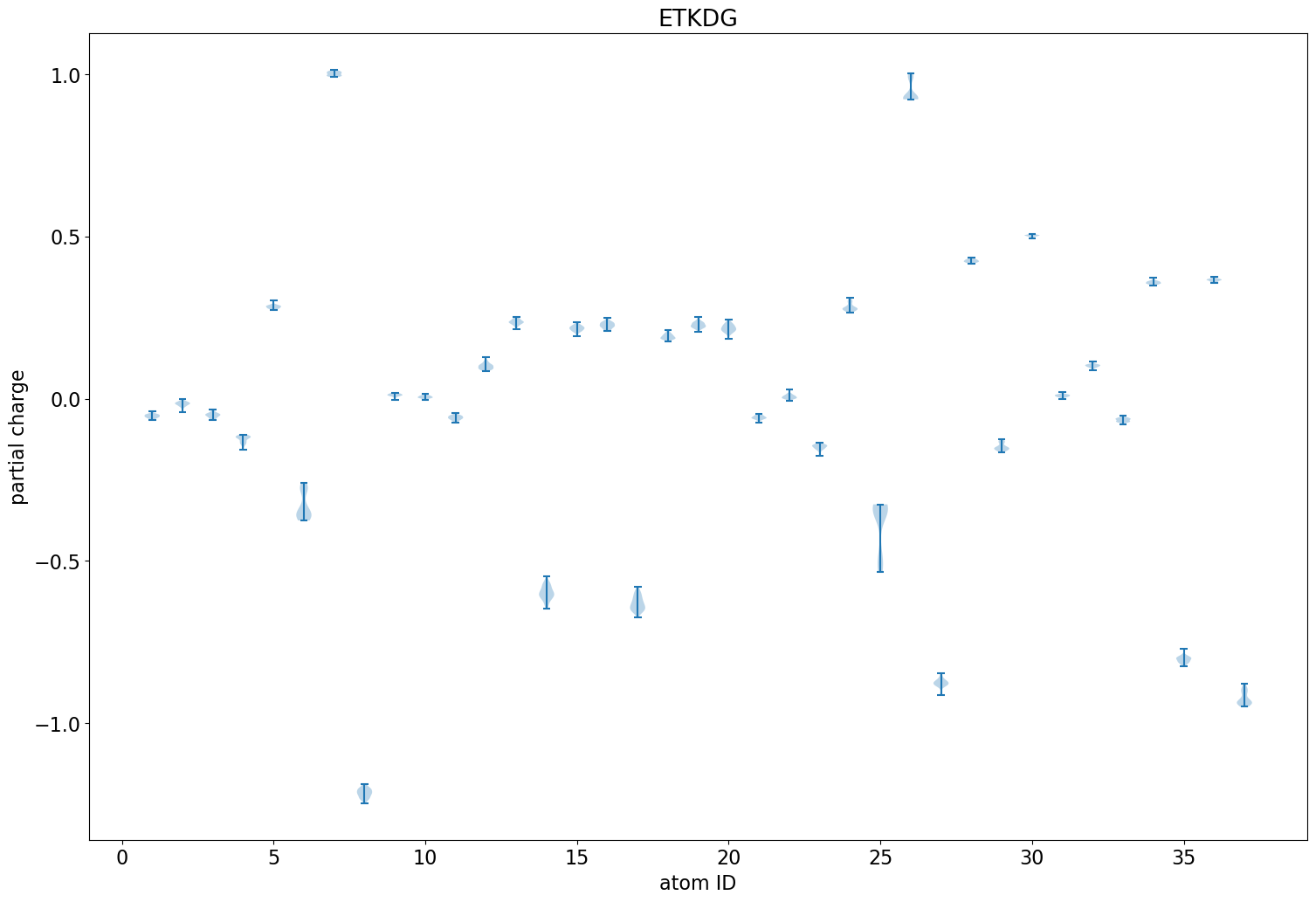
The rest of this blog post is going to show a straightforward analysis one can do with the eHT results: looking at the variability of the QM charges across the different conformers.
def runEHT(mh):""" convenience function to run an eHT calculation on all of a molecule's conformers returns a list of the results structures as well as a list of lists containing the charges on each atom in each conformer: chgs[atomId][confId] to use this """ eres = [] charges = [[] for x inrange(mh.GetNumHeavyAtoms())]for cid inrange(mh.GetNumConformers()): passed,res = rdEHTTools.RunMol(mh,confId=cid) eres.append(res)ifnot passed:raiseValueError("eHT failed") hvyIdx =0 echgs = res.GetAtomicCharges()for atom in mh.GetAtoms():if atom.GetAtomicNum()==1:continue charges[hvyIdx].append(echgs[atom.GetIdx()]) hvyIdx+=1return (eres,charges)
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
The difference here is the relative orientations of the two aromatic rings that N24 is bonded to. In the first case (more negative N), the rings are perpendicular to each other. In the second (less negative N), they are parallel.
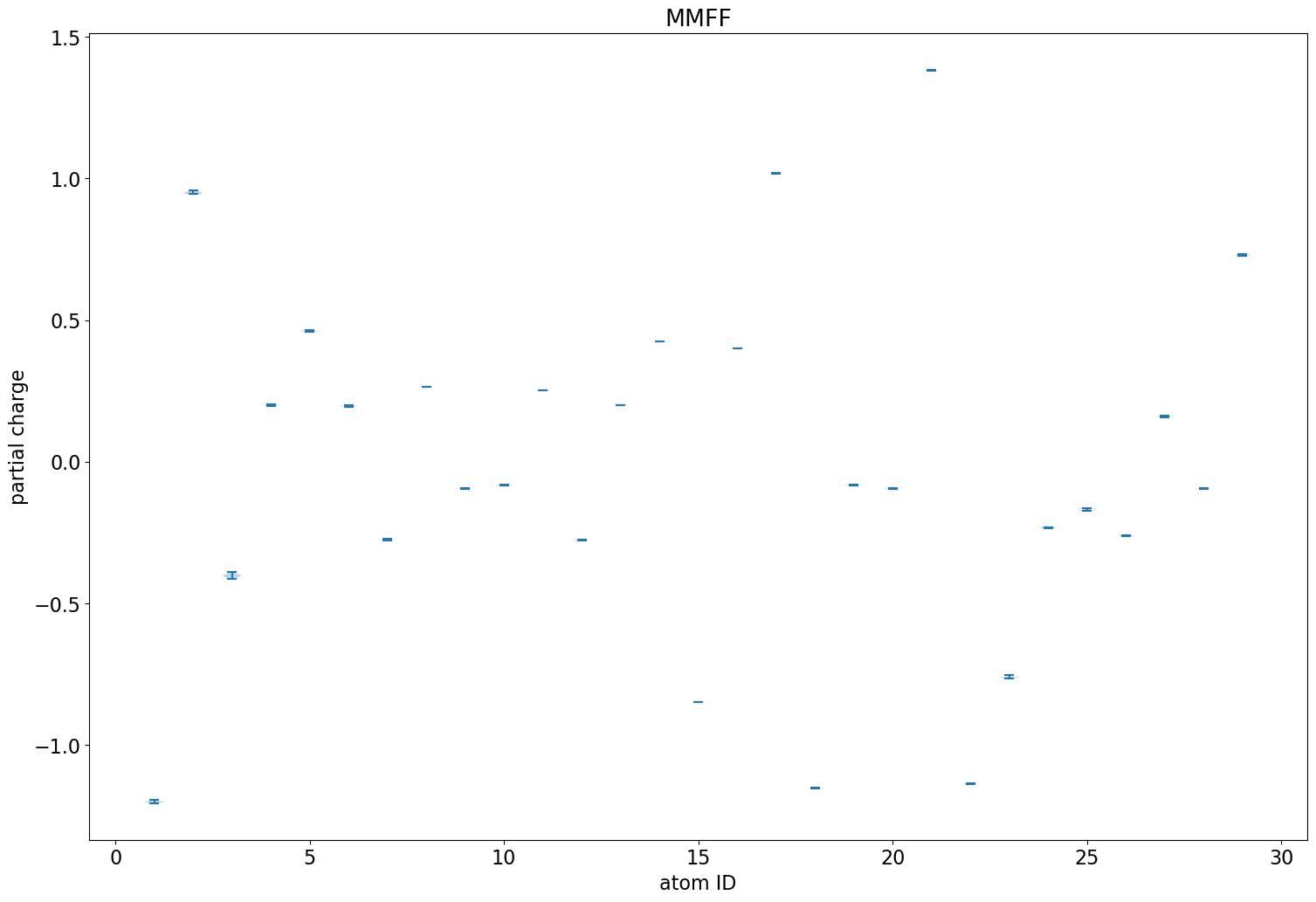
What if we do an MMFF minimization of each conformer?
Less variability than we had with the ETKDG conformers of imatinib, but there is still some. This almost entirely vanishes when using the MMFF-optimized conformers:
40.3 ms ± 391 μs per loop (mean ± std. dev. of 7 runs, 10 loops each)
Do the eHT calculation:
%timeit _ = rdEHTTools.RunMol(tempM)
19.4 ms ± 51.5 μs per loop (mean ± std. dev. of 7 runs, 10 loops each)
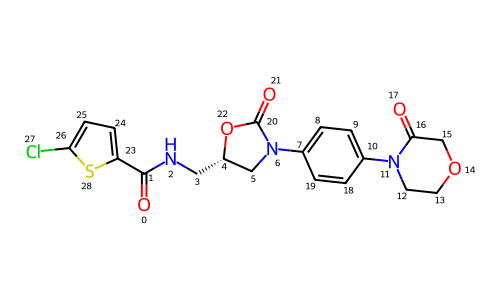
So we can generate a conformer for rivaroxaban, MMFF minimize it, and then do the eHT calculation to get, for example, partial charges (remember these don’t seem to change that much across MMFF conformers) in about 60ms. Not bad!