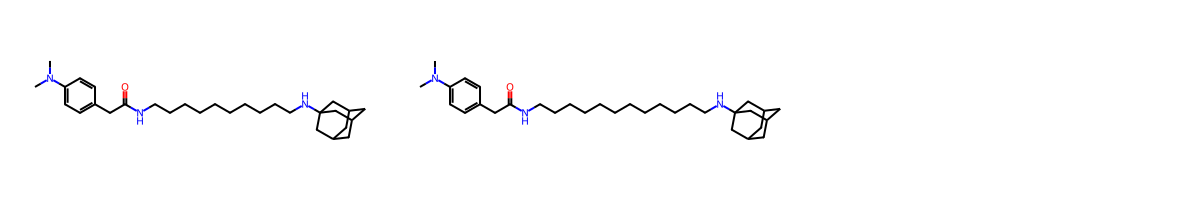
MCES SMARTS : CN(-C)-c1:c:c:c(-CC(=O)-NCCCCCCCCCC):c:c:1.NC12CC3CC(-C1)-CC(-C2)-C3
Matching Bonds : [(0, 0), (1, 1), (2, 2), (3, 3), (4, 4), (5, 5), (6, 6), (7, 7), (8, 8), (9, 9), (10, 10), (11, 11), (12, 12), (13, 13), (14, 14), (15, 15), (16, 16), (17, 17), (18, 18), (19, 19), (21, 23), (22, 24), (23, 25), (24, 26), (25, 27), (26, 28), (27, 29), (28, 30), (29, 31), (30, 32), (31, 33), (32, 34), (33, 35), (34, 36), (35, 37), (36, 38)]
Matching Atoms : [(0, 0), (1, 1), (2, 2), (3, 3), (4, 4), (5, 5), (6, 6), (7, 7), (8, 8), (9, 9), (10, 10), (11, 11), (12, 12), (13, 13), (14, 14), (15, 15), (16, 16), (17, 17), (18, 18), (19, 19), (20, 20), (21, 23), (22, 24), (23, 25), (24, 26), (25, 27), (26, 28), (27, 29), (28, 30), (29, 31), (30, 32), (31, 33), (32, 34), (33, 35)]