from rdkit import Chem
from rdkit.Chem import rdFMCS
from rdkit.Chem.Draw import IPythonConsole, MolsToGridImageNew MCS features in 2023.09.1
release
tutorial
guest post
A big collection of fixes and features.
This guest post by Paolo Tosco (GitHub: @ptosco), one of the RDKit core maintainers, is the fourth of a few posts covering some of the new or improved features in the 2023.09.1 RDKit release.
A PR of mine dealing with MCS code clean up, refactoring and fixing open bugs has been open for one year or so.
The growing sense of guilt, together with new and more exciting bugs being found by myself and others during this year, prompted me to take it up again.
Eventually, I managed to complete the PR, which fixed all outstanding MCS-related bugs at the time of PR submission.
The PR was merged with no changes.
A quick glance at the PR will tell you that this was not necessarily due to my impeccable code style.
In addition to several bug fixes, the PR introduces significant performance improvements, particularly for difficult MCS cases involving the CompleteRingsOnly flag, which now run in finite time rather than just hanging and slowly consuming all of the available memory as they used to.

Here I would like to showcase the new features introduced as part of this PR, which relate to requests I had from colleagues as well as from members of the community.
Custom Python AtomCompare and BondCompare classes
Actually I have already implemented this feature some time ago, but I am not sure how many users are aware of it; therefore, in the following you will find a quick refresher.
Let’s assume that for some reason we would like to implement a custom MCS criterion that will ignore elements and bond orders/aromaticity in ring systems, while enforcing element and bond order consideration in non-ring portions of the molecules.
This is not something which is readily available among the built-in AtomCompare and BondCompare options; therefore, we will need custom subclasses, respectively derived from AtomCompare and BondCompare classes.
mols = [Chem.MolFromSmiles(smi) for smi in """C[C@H]1COC2=C3N1C=C(C(=O)C3=CC(=C2N4CCN(CC4)C)F)C(=O)O
C1[C@@H]2[C@@H](C2N)CN1C3=C(C=C4C(=O)C(=CN(C4=N3)C5=C(C=C(C=C5)F)F)C(=O)O)F
CN(C)CC[C@@](C1=CC=CC2=CC=CC=C21)([C@H](C3=CC=CC=C3)C4=C(N=C5C=CC(=CC5=C4)Br)OC)O
""".split()]MolsToGridImage(mols, subImgSize=(300, 200))
WIth this rather standard set of parameters, which enforces that ring bonds should only match ring bonds and that rings should be complete, the MCS is 3-methylpyridine:
params = rdFMCS.MCSParameters()
params.AtomTyper = rdFMCS.AtomCompare.CompareElements
params.BondTyper = rdFMCS.BondCompare.CompareOrder
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = Truemcs = rdFMCS.FindMCS(mols, params)MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(mcs.queryMol) for m in mols])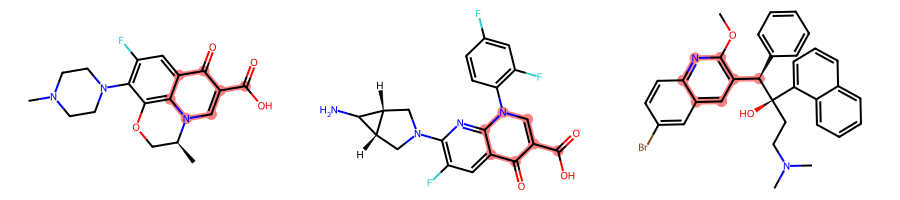
class CompareOrderOutsideRings(rdFMCS.MCSBondCompare):
def __call__(self, p, mol1, bond1, mol2, bond2):
b1 = mol1.GetBondWithIdx(bond1)
b2 = mol2.GetBondWithIdx(bond2)
if (b1.IsInRing() and b2.IsInRing()) or (b1.GetBondType() == b2.GetBondType()):
if (p.MatchStereo and not self.CheckBondStereo(p, mol1, bond1, mol2, bond2)):
return False
if p.RingMatchesRingOnly:
return self.CheckBondRingMatch(p, mol1, bond1, mol2, bond2)
return True
return Falseclass CompareElementsOutsideRings(rdFMCS.MCSAtomCompare):
def __call__(self, p, mol1, atom1, mol2, atom2):
a1 = mol1.GetAtomWithIdx(atom1)
a2 = mol2.GetAtomWithIdx(atom2)
if (a1.GetAtomicNum() != a2.GetAtomicNum()) and not (a1.IsInRing() and a2.IsInRing()):
return False
if (p.MatchChiralTag and not self.CheckAtomChirality(p, mol1, atom1, mol2, atom2)):
return False
if p.RingMatchesRingOnly:
return self.CheckAtomRingMatch(p, mol1, atom1, mol2, atom2)
return TrueBy using our more lenient atom/bond comparison criterion, while keeping the constraint that ring atoms should only match ring atoms and that rings should be complete, we retrieve a larger MCS:
params = rdFMCS.MCSParameters()
params.AtomTyper = CompareElementsOutsideRings()
params.BondTyper = CompareOrderOutsideRings()
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = Truemcs = rdFMCS.FindMCS(mols, params)MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(mcs.queryMol) for m in mols])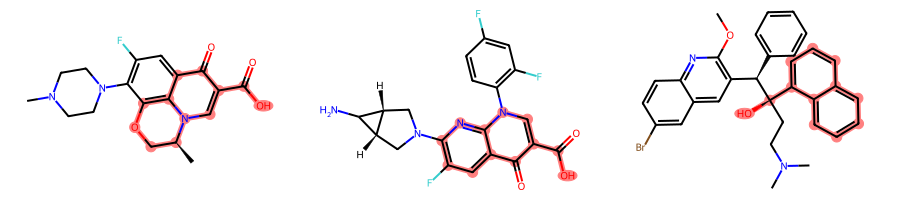
Note that the first molecule can actually match two equivalent ring systems:
[m.GetSubstructMatches(mcs.queryMol) for m in mols][((1, 2, 6, 3, 7, 4, 8, 5, 9, 23, 11, 25),
(5, 4, 6, 14, 7, 13, 8, 12, 9, 23, 11, 25)),
((9, 8, 10, 7, 11, 17, 13, 16, 14, 26, 15, 28),),
((9, 10, 8, 11, 7, 12, 6, 13, 15, 5, 14, 36),)]The fact that we instantiate custom compare classes rather than custom compare functions allows to put some non-molecule specific (i.e., static) data that we may need for the comparison in addition to the MCSParameters that get passed to the __call__ method by the MCS code.
We may access this data stored on the class instance inside the __call__ method.
For example, let’s assume that we want to enforce that aromatic only matches aromatic based on a user-specified setting.
All we need to do is modify our CompareOrderOutsideRings as follows:
class CompareOrderOutsideRings(rdFMCS.MCSBondCompare):
def __init__(self, aromatic_match_aromatic=False):
super().__init__()
self.aromatic_match_aromatic = aromatic_match_aromatic
def __call__(self, p, mol1, bond1, mol2, bond2):
b1 = mol1.GetBondWithIdx(bond1)
b2 = mol2.GetBondWithIdx(bond2)
if ((b1.IsInRing() and b2.IsInRing()
and (not self.aromatic_match_aromatic or not (b1.GetIsAromatic() ^ b2.IsInRing())))
or b1.GetBondType() == b2.GetBondType()):
if (p.MatchStereo and not self.CheckBondStereo(p, mol1, bond1, mol2, bond2)):
return False
if p.RingMatchesRingOnly:
return self.CheckBondRingMatch(p, mol1, bond1, mol2, bond2)
return True
return FalseIf we instantiate the CompareOrderOutsideRings class without passing the aromatic_match_aromatic optional parameter, which defaults to False, we will get the same results as before:
params = rdFMCS.MCSParameters()
params.AtomTyper = CompareElementsOutsideRings()
params.BondTyper = CompareOrderOutsideRings()
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = Truemcs = rdFMCS.FindMCS(mols, params)MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(mcs.queryMol) for m in mols])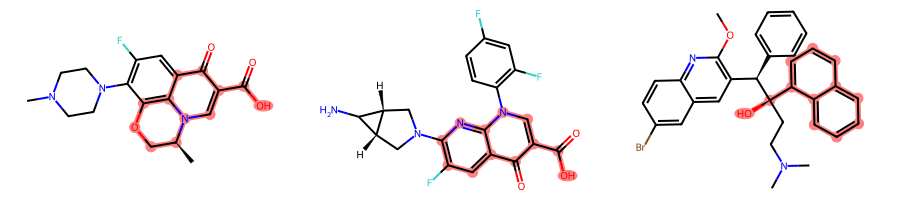
[m.GetSubstructMatches(mcs.queryMol) for m in mols][((1, 2, 6, 3, 7, 4, 8, 5, 9, 23, 11, 25),
(5, 4, 6, 14, 7, 13, 8, 12, 9, 23, 11, 25)),
((9, 8, 10, 7, 11, 17, 13, 16, 14, 26, 15, 28),),
((9, 10, 8, 11, 7, 12, 6, 13, 15, 5, 14, 36),)]However, if we instantiate the CompareOrderOutsideRings class setting the aromatic_match_aromatic optional parameter to True, the first molecule will match only on the quinoline ring system:
params = rdFMCS.MCSParameters()
params.AtomTyper = CompareElementsOutsideRings()
params.BondTyper = CompareOrderOutsideRings(True)
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = Truemcs = rdFMCS.FindMCS(mols, params)MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(mcs.queryMol) for m in mols])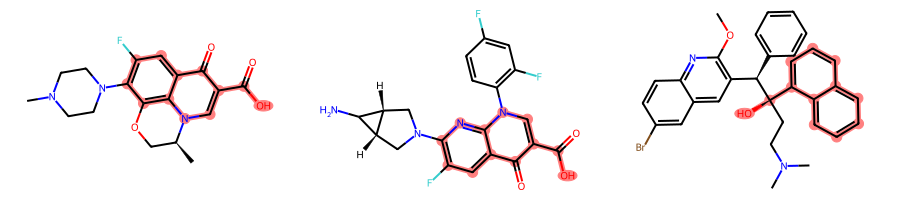
[m.GetSubstructMatches(mcs.queryMol) for m in mols][((4, 5, 14, 6, 13, 7, 12, 8, 11, 9, 23, 25),),
((8, 9, 7, 10, 17, 11, 16, 13, 15, 14, 26, 28),),
((10, 9, 11, 8, 12, 7, 13, 6, 14, 15, 5, 36),)]Retrieve all degenerate MCSs
It may happen that there are multiple degenerate MCSs (i.e., with the same size) across a set of molecules.
Before my PR, the MCS algorithm implemented in RDKit would return only one of them, but which one would be rather unpredictable, as it depends on atom ordering in the molecules.
This topic was raised during an RDKit UGM 2-3 years ago, but I did not realize its importance until one of my colleagues was bitten by the degenerate MCS issue.
Now there is an optional StoreAll parameter, which defaults to False, which if set to True will trigger the storage of all degenerate MCSs in an array.
See below for an example.
mols = [Chem.MolFromSmiles(smi) for smi in [
"Nc1ccc(cc1)C-Cc1c(N)cccc1",
"Nc1ccc(cc1)C=Cc1c(N)cccc1"]
]MolsToGridImage(mols, subImgSize=(300, 200))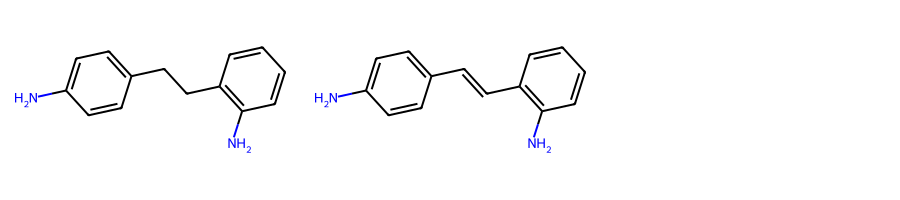
You can see at a glance that two degenerate MCSs can be found between the above molecules, i.e., o-methylaniline and p-methylaniline.
However, by default rdFMCS will only find one, in this case the para isomer:
mcs = rdFMCS.FindMCS(mols)mcs.numAtoms, mcs.numBonds(8, 8)mcs.queryMol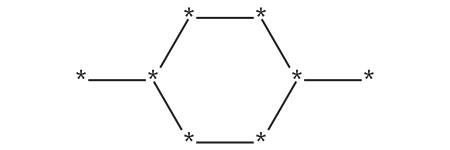
MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(mcs.queryMol) for m in mols])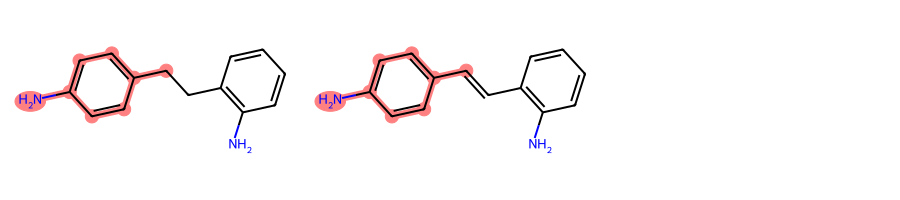
params = rdFMCS.MCSParameters()
params.StoreAll = Truemcs = rdFMCS.FindMCS(mols, params)The number of atoms and bonds that constitute the MCS, as expected, are the same as before, since the MCSs are degenerate:
mcs.numAtoms, mcs.numBonds(8, 8)However, this time mcs.queryMol and mcs.smartsString are None and '', respectively.
This is by design, as with StoreAll we need to be able to potentially access multiple MCSs, rather than only one as is the case for the MCSResult.queryMol and MCSResult.smartsString properties.
The property that allows to browse all degenerate MCSs is MCSResult.degenerateSmartsQueryMolDict, which is a dict:
mcs.queryMolmcs.smartsString''mcs.degenerateSmartsQueryMolDict{'[#6]-[#6]1:[#6]:[#6]:[#6]:[#6]:[#6]:1-[#7]': <rdkit.Chem.rdchem.Mol at 0x21594654c10>,
'[#7]-[#6]1:[#6]:[#6]:[#6](:[#6]:[#6]:1)-[#6]': <rdkit.Chem.rdchem.Mol at 0x215946550e0>}Below I show all degenerate MCSs found, labelled with their respective SMARTS pattern:
mols_highlights_smarts_list = [[mols, [m.GetSubstructMatch(query) for m in mols], [smarts for _ in mols]] for smarts, query in mcs.degenerateSmartsQueryMolDict.items()]mols_to_display, highlight_atom_lists, legends = [sum(item, []) for item in map(list, list(zip(*mols_highlights_smarts_list)))]MolsToGridImage(mols_to_display, molsPerRow=2, subImgSize=(300, 200), highlightAtomLists=highlight_atom_lists, legends=legends)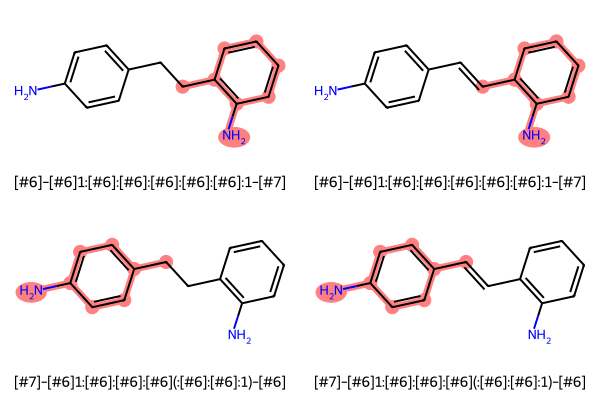
FinalMatchChecker
MCSParameters.FinalMatchChecker allows to implement a final check on a growing MCS to determine whether it should be accepted (and eventually be allowed to grow further, if possible) or discarded.
Just to clarify a bit, the MCS algorithm already implements some built-in, optional final match checkers, e.g. to check: * stereochemistry, in case MCSParameters.AtomCompareParameters.MatchChiralTag is set to True * ring fusion, in case MCSParameters.BondCompareParameters.MatchFusedRings is set to True
By implementing a custom class derived from rdFMCS.MCSFinalMatchCheck you may enforce your own check.
For example, the following class will prevent any MCS containing nitrogen from growing further, causing it to be discarded straightaway:
class FinalMatchCheckNoNitrogen(rdFMCS.MCSFinalMatchCheck):
def __call__(self, mol1, mol2, atom_idx_match, bond_idx_match, params):
return not any(mol1.GetAtomWithIdx(mol1_atom_idx).GetAtomicNum() == 7
or mol2.GetAtomWithIdx(mol2_atom_idx).GetAtomicNum() == 7
for mol1_atom_idx, mol2_atom_idx in atom_idx_match)Taking again as an example the two molecules from above, if we blacklist nitrogen we would expect the MCS to be toluene.
mols = [Chem.MolFromSmiles(smi) for smi in [
"Nc1ccc(cc1)C-Cc1c(N)cccc1",
"Nc1ccc(cc1)C=Cc1c(N)cccc1"]
]MolsToGridImage(mols, subImgSize=(300, 200))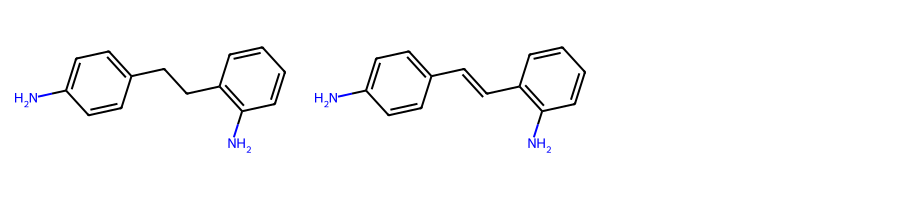
params = rdFMCS.MCSParameters()
params.FinalMatchChecker = FinalMatchCheckNoNitrogen()mcs = rdFMCS.FindMCS(mols, params)The number of atoms and bonds that constitute the MCS, as expected, are the same as before, since the MCSs are degenerate:
mcs.numAtoms, mcs.numBonds(7, 7)And indeed, toluene is what we get:
mcs.queryMol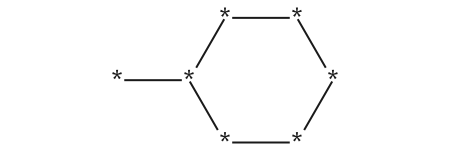
mcs.smartsString'[#6]1:[#6]:[#6]:[#6](:[#6]:[#6]:1)-[#6]'MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(mcs.queryMol) for m in mols])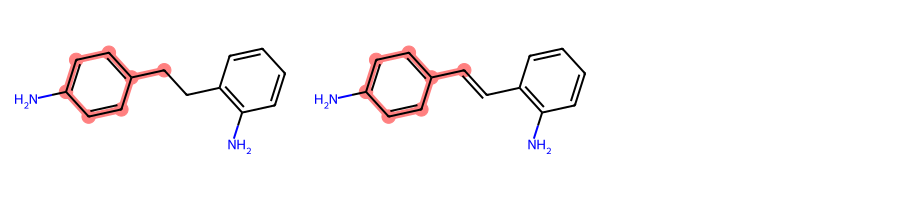
ShouldAcceptMCS
MCSParameters.ShouldAcceptMCS allows to implement a user-defined acceptance criterion on a complete MCS (which is already fully grown and potentially ready to be returned as result).
If the complete NCS fails the check, it will be discarded and not returned to the user.
A quite specific use case that we recently had internally was to find an MCS between two molecules, one of which containing a dummy atom.
We needed to enforce that, for the molecule containing the dummy atom, this dummy atom was actually part of the MCS.
We did this by using a custom TerminalDummyMatchesAnyNonTerminalAtom AtomCompare subclass and a custom EnforceDummyAcceptance MCSAcceptance subclass; see below.
class TerminalDummyMatchesAnyNonTerminalAtom(rdFMCS.MCSAtomCompare):
def __call__(self, p, mol1, atom1, mol2, atom2):
a1 = mol1.GetAtomWithIdx(atom1)
a2 = mol2.GetAtomWithIdx(atom2)
a1_is_dummy = (a1.GetAtomicNum() == 0)
a2_is_dummy = (a2.GetAtomicNum() == 0)
if a1_is_dummy ^ a2_is_dummy:
atoms = (a1, a2)
dummy_atom_idx = 0 if a1_is_dummy else 1
other_atom_idx = 1 - dummy_atom_idx
return atoms[dummy_atom_idx].GetDegree() == 1 and atoms[other_atom_idx].GetDegree() > 1
if a1.GetAtomicNum() != a2.GetAtomicNum():
return False
return self.CheckAtomRingMatch(p, mol1, atom1, mol2, atom2)class EnforceDummyAcceptance(rdFMCS.MCSAcceptance):
def __call__(self, mol1, mol2, atom_idx_match, bond_idx_match, params):
for mol1_atom_idx, mol2_atom_idx in atom_idx_match:
if mol1.GetAtomWithIdx(mol1_atom_idx).GetAtomicNum() == 0:
return True
if mol2.GetAtomWithIdx(mol2_atom_idx).GetAtomicNum() == 0:
return True
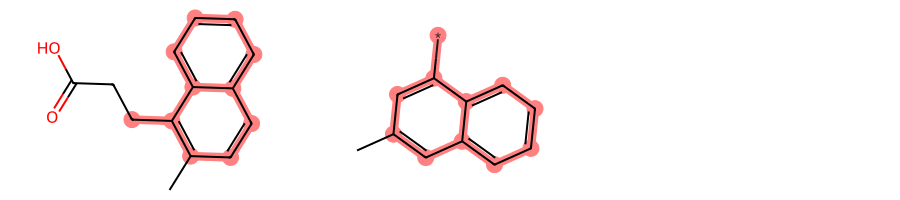
return Falsemols = [
Chem.MolFromSmiles(smi) for smi in [
"OC(=O)CCc1c(C)ccc2ccccc12",
"c1(C)cc(cccc2)c2c(*)c1"
]
]MolsToGridImage(mols, subImgSize=(300, 200))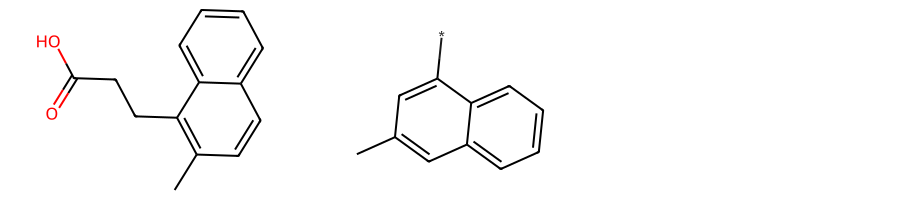
If we set neither a custom AtomCompare class nor a custom MCSAcceptance but rather only request BondCompareParameters.RingMatchesRingOnly and BondCompareParameters.CompleteRingsOnly and set StoreAll to True, the only MCS found will be 2-naphthalene:
params = rdFMCS.MCSParameters()
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = True
params.StoreAll = Truemcs = rdFMCS.FindMCS(mols, params)mcs.numAtoms, mcs.numBonds(11, 12)len(mcs.degenerateSmartsQueryMolDict)1smarts_string, query_mol = tuple(mcs.degenerateSmartsQueryMolDict.items())[0]smarts_string'[#6]1(-&!@[#6]):&@[#6]:&@[#6]2:&@[#6]:&@[#6]:&@[#6]:&@[#6]:&@[#6]:&@2:&@[#6]:&@[#6]:&@1'MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(query_mol) for m in mols])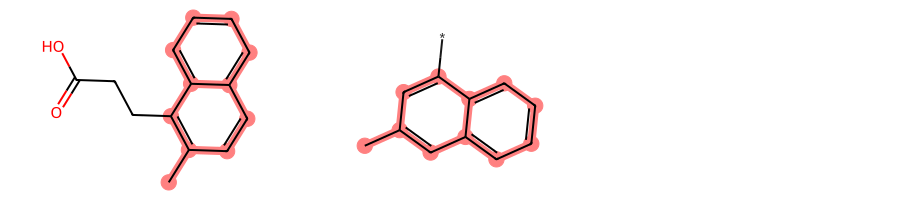
Instead, if in addiiton to the above we specify the custom TerminalDummyMatchesAnyNonTerminalAtom AtomCompare subclass allowing dummy atoms to match any other atom, we will find two degenerate MCSs:
params = rdFMCS.MCSParameters()
params.AtomTyper = TerminalDummyMatchesAnyNonTerminalAtom()
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = True
params.StoreAll = Truemcs = rdFMCS.FindMCS(mols, params)mcs.numAtoms, mcs.numBonds(11, 12)len(mcs.degenerateSmartsQueryMolDict)2mcs.degenerateSmartsQueryMolDict{'[#6]1(-&!@[#6]):&@[#6]:&@[#6]:&@[#6]2:&@[#6]:&@[#6]:&@[#6]:&@[#6]:&@[#6]:&@2:&@[#6]:&@1': <rdkit.Chem.rdchem.Mol at 0x215946566c0>,
'[#6]1:&@[#6]:&@[#6]2:&@[#6](:&@[#6](-&!@[#0,#6]):&@[#6]:&@1):&@[#6]:&@[#6]:&@[#6]:&@[#6]:&@2': <rdkit.Chem.rdchem.Mol at 0x21594656810>}Below I display I display all degenerate MCSs found, labelled with their respective SMARTS pattern:
mols_highlights_smarts_list = [[mols, [m.GetSubstructMatch(query) for m in mols], [smarts for _ in mols]] for smarts, query in mcs.degenerateSmartsQueryMolDict.items()]mols_to_display, highlight_atom_lists, legends = [sum(item, []) for item in map(list, list(zip(*mols_highlights_smarts_list)))]MolsToGridImage(mols_to_display, molsPerRow=2, subImgSize=(300, 200), highlightAtomLists=highlight_atom_lists, legends=legends)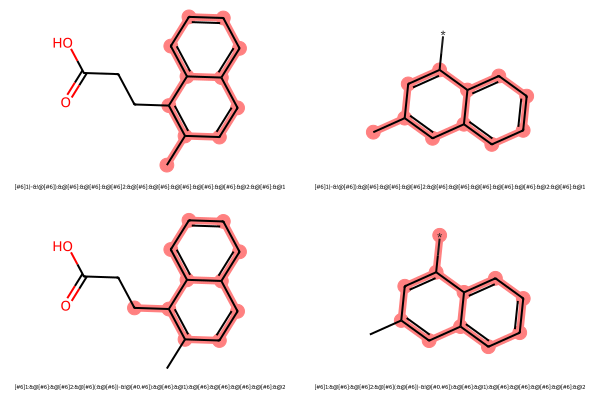
As expected, since we allowed terminal dummies to match any non-terminal atom, we get both 1- and 2-naphthalene as degenerate MCSs.
Finally, if in addition to the above we also use EnforceDummyAcceptance as custom MCSAcceptance criterion enforcing that the dummy atom should be part of the MCS if possible, we will again get a single MCS, i.e. 1-naphthalene as it is the only one including the dummy atom:
params = rdFMCS.MCSParameters()
params.AtomTyper = TerminalDummyMatchesAnyNonTerminalAtom()
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = True
params.ShouldAcceptMCS = EnforceDummyAcceptance()
params.StoreAll = Truemcs = rdFMCS.FindMCS(mols, params)mcs.numAtoms, mcs.numBonds(11, 12)len(mcs.degenerateSmartsQueryMolDict)1smarts_string, query_mol = tuple(mcs.degenerateSmartsQueryMolDict.items())[0]smarts_string'[#6]1:&@[#6]:&@[#6]2:&@[#6](:&@[#6](-&!@[#0,#6]):&@[#6]:&@1):&@[#6]:&@[#6]:&@[#6]:&@[#6]:&@2'MolsToGridImage(mols, subImgSize=(300, 200), highlightAtomLists=[m.GetSubstructMatch(query_mol) for m in mols])