from rdkit import Chem
from rdkit.Chem import rdDistGeom
from rdkit.Chem import rdDepictor
rdDepictor.SetPreferCoordGen(True)
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.ipython_3d = True
import rdkit
print(rdkit.__version__)2023.03.1When the RDKit generates conformers for a molecule it performs a number of checks along the way to make sure that the atomic coordinates are reasonable/correct. When one of these checks fails, the current conformer is abandoned and the coordinate generation process starts from scratch. The maximum number of times this restart can happen is controlled by the maxIterations parameter: after maxIterations restarts the conformer generation fails. If you’ve called EmbedMolecule(), asking for a single conformer, you’ll get a conformation ID of -1 to mark this failure. If you’ve asked for multiple conformers, you won’t get as many as you requested.
One of the changes for the 2023.03.1 release of the RDKit was the addition of an option to track which checks are failing during conformer generation. This can help understand what problems were encountered and to understand why conformer generation is not generating the expected results. Since, as is often the case, the RDKit’s capabilities are ahead of its documentation, this short blog post will explain how to use this functionality and interpret the results.
A general overview of the conformer generation process can be found in the documentation.
from rdkit import Chem
from rdkit.Chem import rdDistGeom
from rdkit.Chem import rdDepictor
rdDepictor.SetPreferCoordGen(True)
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.ipython_3d = True
import rdkit
print(rdkit.__version__)2023.03.1Molecules containing chiral centers in bridged rings can cause problems, so let’s start with a norbornane derivative:
m = Chem.MolFromSmiles('C1C[C@]2(F)CC[C@@]1(Cl)C2')
m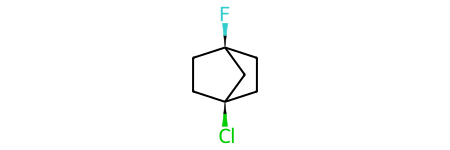
mh = Chem.AddHs(m)
ps = rdDistGeom.ETKDGv3()
ps.randomSeed = 0xf00d
rdDistGeom.EmbedMolecule(mh,ps)0mhYou appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
Now invert one of the stereocenters to create the stereoisomer which can’t actually exist as a stable structure. Imagine inverting one of the chiral centers in the above molecule and you will see the problem: we’d have to point the attached halogen into the center of the cage.
bad_m = Chem.MolFromSmiles('C1C[C@]2(F)CC[C@]1(Cl)C2')
bad_m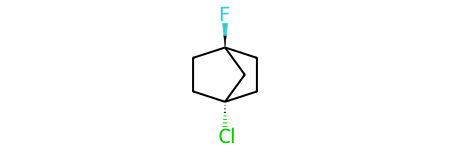
Try creating a conformer for that:
bad_mh = Chem.AddHs(bad_m)
ps = rdDistGeom.ETKDGv3()
ps.randomSeed = 0xf00d
rdDistGeom.EmbedMolecule(bad_mh,ps)-1The new functionality allows us to keep track of why the embedding failed:
ps.trackFailures = True
print(rdDistGeom.EmbedMolecule(bad_mh,ps))
counts = ps.GetFailureCounts()
counts-1(7, 11, 30, 0, 0, 0, 142, 0, 0, 0)Here’s a quick loop over all the failure causes to see where the problems were:
def print_failure_causes(counts):
for i,k in enumerate(rdDistGeom.EmbedFailureCauses.names):
print(k,counts[i])
# in v2022.03.1 two names are missing from `rdDistGeom.EmbedFailureCauses`:
print('LINEAR_DOUBLE_BOND',counts[i+1])
print('BAD_DOUBLE_BOND_STEREO',counts[i+2]) print_failure_causes(counts)INITIAL_COORDS 7
FIRST_MINIMIZATION 11
CHECK_TETRAHEDRAL_CENTERS 30
CHECK_CHIRAL_CENTERS 0
MINIMIZE_FOURTH_DIMENSION 0
ETK_MINIMIZATION 0
FINAL_CHIRAL_BOUNDS 142
FINAL_CENTER_IN_VOLUME 0
LINEAR_DOUBLE_BOND 0
BAD_DOUBLE_BOND_STEREO 0Here’s what the individual failures mean:
INITIAL_COORDS: generation of the initial coordinates from the random distance matrix (default) or from a set of random coordinates (when using random coordinate embedding) failed.FIRST_MINIMIZATION: the initial optimization of the atom positions using the distance-geometry force field failed to produce a low-enough energy conformer. The check here has thresholds for both average energy per atom and the individual atom energies. I’m not providing the threshold values here since the energies from the distance-geometry force field are not physically meaningful - the threshold values are not interpretable.CHECK_TETRAHEDRAL_CENTERS: at least one tetrahedral C and N centers either has a volume around it which is too small or is outside the volume defined by its neighborsMINIMIZE_FOURTH_DIMENSION: the minmization to force the values of the fourth-dimensional component of each atom position failedETK_MINIMIZATION: after the minimization with the ET and/or K terms, at least one atom which should have been planar was notFINAL_CHIRAL_BOUNDS: the neighborhood of an atom with specified chirality was too distorted (it violated distance constraints)FINAL_CENTER_IN_VOLUME: an atom with specified chirality was outside of the volume defined by its neighborsLINEAR_DOUBLE_BOND: one of the end atoms of a double bond had a linear geometryBAD_DOUBLE_BOND_STEREO: the stereochemistry of a double bond with specified stereochemistry was wrong in the generated conformerJust for comparison, the starting molecule had no failures:
ps.trackFailures = True
print(rdDistGeom.EmbedMolecule(mh,ps))
counts = ps.GetFailureCounts()
counts0(0, 0, 0, 0, 0, 0, 0, 0, 0, 0)Let’s try a more complicated molecule from ChEMBL; specified bond stereochemistry in macrocycles can also make conformer generation challenging.
m = Chem.MolFromSmiles(r'COc1cc(O)c2CSC[C@H](\N=C(/S)\CCC[C@@H](CO)OC(=O)c2c1C)c3onc(C)n3')
m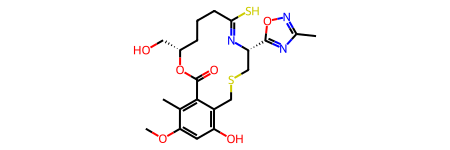
mh = Chem.AddHs(m)
ps = rdDistGeom.ETKDGv3()
ps.randomSeed = 0xbadd06
ps.trackFailures = True
print(rdDistGeom.EmbedMolecule(mh,ps))
print_failure_causes(ps.GetFailureCounts())0
INITIAL_COORDS 2
FIRST_MINIMIZATION 0
CHECK_TETRAHEDRAL_CENTERS 0
CHECK_CHIRAL_CENTERS 0
MINIMIZE_FOURTH_DIMENSION 0
ETK_MINIMIZATION 0
FINAL_CHIRAL_BOUNDS 0
FINAL_CENTER_IN_VOLUME 0
LINEAR_DOUBLE_BOND 3
BAD_DOUBLE_BOND_STEREO 4Here we did get a conformer, but there were some rejected conformers along the way. The double-bond-related failures indicate that the code had some problems with the ring double bond with specified stereochemistry.