Quite a while ago I did a blog post showing how to use the Python function AllChem.ConstrainedEmbed() to generate conformers where the positions of a set of atoms are constrained to match the coordinates of a template molecule. More recently a question came up on the mailing list about how to use the core embedding functionality that lies underneath AllChem.ConstrainedEmbed(); this short post will look at that.
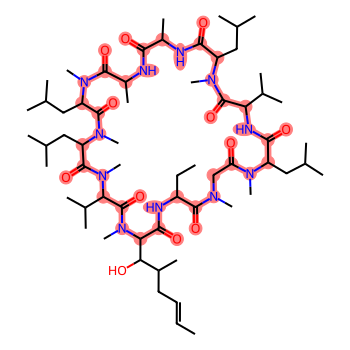
We’re going to work with cyclosporine here, since it’s delightfully complicated, which makes it fun. :-) In this case we’re going to ignore atomic stereochemistry in order to speed the conformer generation up (the RDKit tends to take a while to generate conformers for molecules with a large number of stereocenters).
We’ll also define the macrocylce as the core; this is what we’re going to use to provide constraints.
m = Chem.MolFromSmiles('C/C=C/CC(C)C(O)C1C(=O)NC(CC)C(=O)N(C)CC(=O)N(C)C(CC(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(CC(C)C)C(=O)NC(C)C(=O)NC(C)C(=O)N(C)C(CC(C)C)C(=O)N(C)C(CC(C)C)C(=O)N(C)C(C(C)C)C(=O)N1C')rdDepictor.Compute2DCoords(m)core = Chem.MolFromSmiles('C1C(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)N1')m.GetSubstructMatch(core)m
Start by generating a conformer for cyclosporine itself.
There’s no reason to expect this RMSD to be anything other than huge: we’re using different conformer of a flexible core and haven’t aligned them to each other.
We can go ahead and do an alignmeent and see how that affects the RMSD:
Again, it’s not surprising that this is a large RMSD: the core is quite flexible and we haven’t constrained it at all.
Adding constraints is the point of this blog post though, so let’s move onto that.
AllChem.EmbedMolecule() can be given constraints for the positions of certain atoms using the coordMap argument, which expects a dictionary that provides a Point3D for each atom that should have a fixed position.
cmap = {newm_match[i]:mh.GetConformer().GetAtomPosition(m_match[i]) for i inrange(len(m_match))}AllChem.EmbedMolecule(newmh,randomSeed=0xf00d,coordMap=cmap)delta2 =0.0for mi,newmi inzip(m_match,newm_match): d = (mh.GetConformer().GetAtomPosition(mi) - newmh.GetConformer().GetAtomPosition(newmi)).LengthSq() delta2 += dprint('core RMSD:',math.sqrt(delta2/len(m_match)))
core RMSD: 2.5231352176127078
Wait… what happened here? Shouldn’t this number be smaller since we introduced constraints? This was the question on the mailing list.
The function AllChem.EmbedMolecule() uses the coordinates provided in coordMap to set elements of the distance bounds matrix that is used to generate conformers (details about the RDKit’s distance-geometry-based conformer generator are in the documentation). This results in conformers where the distances between the atoms in the conformer closely match the corresponding distances in the coordMap.
Note that, because the coordinates are being constrained using the distances between them, you should expect rigid shifts of the core atoms relative to the constraints. This is solveable by aligning the core of the test molecule to the core of the reference:
Note that the output coordinates don’t match the constraint coordinates exactly. This will almost always be the case; they should be close, but some differences are, unforunately, expected due to the nature of the algorithm.
An alternative is to use random coordinate embedding instead of the usual distance-bounds embedding to generate the initial coordinates in the conformer generation. When we do this it’s possible to get exact coordinate matches of the core and no alignment is necessary. Random-coordinate embedding is not the default because the current RDKit implementation tends to be slower than the other approach.
It’s worth taking a look at the conformers of our molecules. We’ll do that using py3Dmol. This code snippet uses a bit of convenience functionality that the RDKit’s IPythonConsole provides, but it also demonstrates how to draw molecules with different colors.
import py3Dmoldef drawit(ms, p=None, confId=-1, removeHs=True,colors=('cyanCarbon','redCarbon','blueCarbon')):if p isNone: p = py3Dmol.view(width=400, height=400) p.removeAllModels()for i,m inenumerate(ms):if removeHs: m = Chem.RemoveHs(m) IPythonConsole.addMolToView(m,p,confId=confId) p.setStyle({'model':-1,}, {'stick':{'colorscheme':colors[i%len(colors)]}}) p.zoomTo()return p.show()
drawit((mh,newmh))
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension: jupyter labextension install jupyterlab_3dmol
Here one molecule is drawn with cyan bonds and the other with red bonds.
Extra: minimize with constraints
from rdkit.Chem import rdForceFieldHelpersmcp = Chem.Mol(newmh)mmffps = rdForceFieldHelpers.MMFFGetMoleculeProperties(mcp)ff = rdForceFieldHelpers.MMFFGetMoleculeForceField(mcp,mmffps)maxIters =10while ff.Minimize(maxIts=1000) and maxIters>0: maxIters -=1
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension: jupyter labextension install jupyterlab_3dmol
That’s messed up the core coordinates… we can fix that by adding some position constraints to the force field:
mcp = Chem.Mol(newmh)mmffps = rdForceFieldHelpers.MMFFGetMoleculeProperties(mcp)ff = rdForceFieldHelpers.MMFFGetMoleculeForceField(mcp,mmffps)for atidx in newm_match: ff.MMFFAddPositionConstraint(atidx,0.05,200)maxIters =10while ff.Minimize(maxIts=1000) and maxIters>0: maxIters -=1
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension: jupyter labextension install jupyterlab_3dmol