Generating molecules from all possible combinations of R groups
Published
March 14, 2022
Recently a couple of customers have asked questions along the lines of: “How do I do an R-group decomposition and then recombine the cores and R groups to create new molecules?” That’s an interesting and useful task which the RDKit has some built-in tools to help with, so I figured I’d do a blog post.
Note: Though I’m doing this blog post using a local build from RDKit master all of the functionality which I demonstrate here is already available in the 2021.09 release series.
Read in the dataset
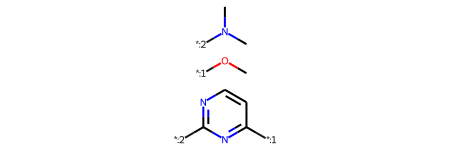
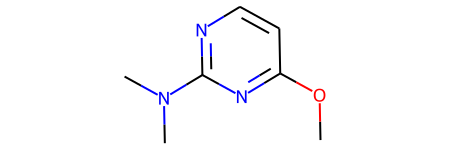
Read in a bunch of molecules from a J Med Chem paper (https://doi.org/10.1021/acs.jmedchem.7b00306 the paper is open access). Here I downloaded the SMILES in the supporting information, sketched the scaffold manually, and then saved the scaffold + molecules to an SD file. The scaffold is the first molecule in the SDF.
ms = [mol for mol in Chem.SDMolSupplier('../data/jm7b00306.sdf')]core = ms[0]ms.pop(0)print(f'There are {len(ms)} molecules')core
There are 31 molecules
scaffold
1
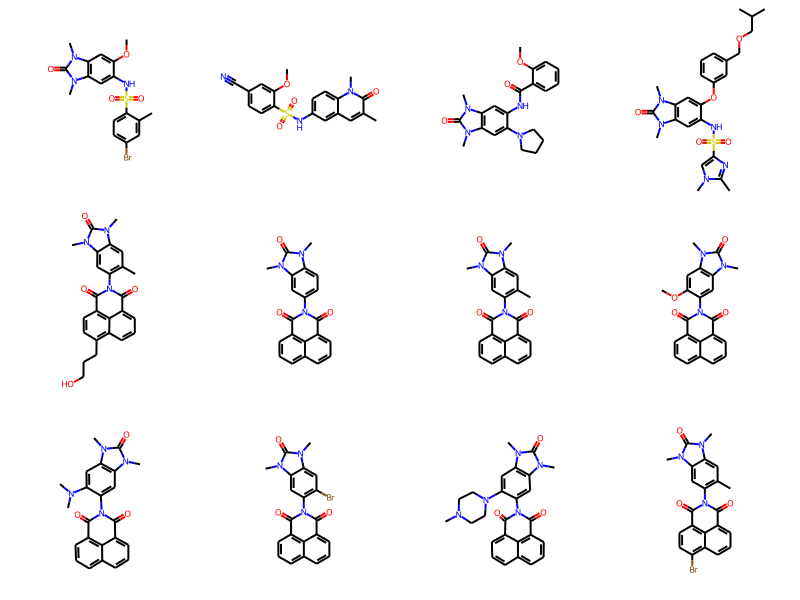
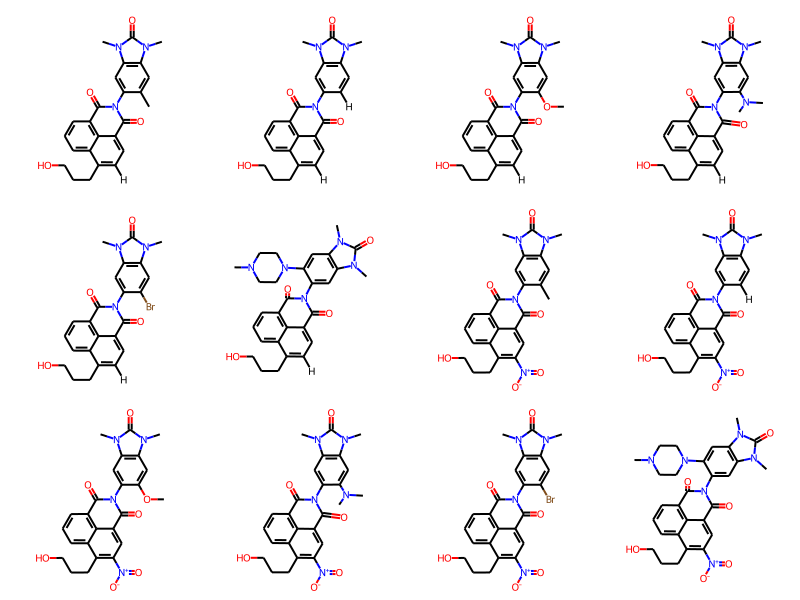
Now let’s look at some of the other molecules
Draw.MolsToGridImage(ms[:12],molsPerRow=4)
Doing the R-group decomposition
The RGD code takes a list of cores to be used along with a list of molecules. It returns a 2-tuple with: 1. a dictionary with the results 2. a list with the indices of the molecules which failed; these are molecules which did not match any of the cores
I’ve blogged about the RGD code before here and here if you want to read more about it. It’s probably time for another post with an update, but those are hopefully already useful.
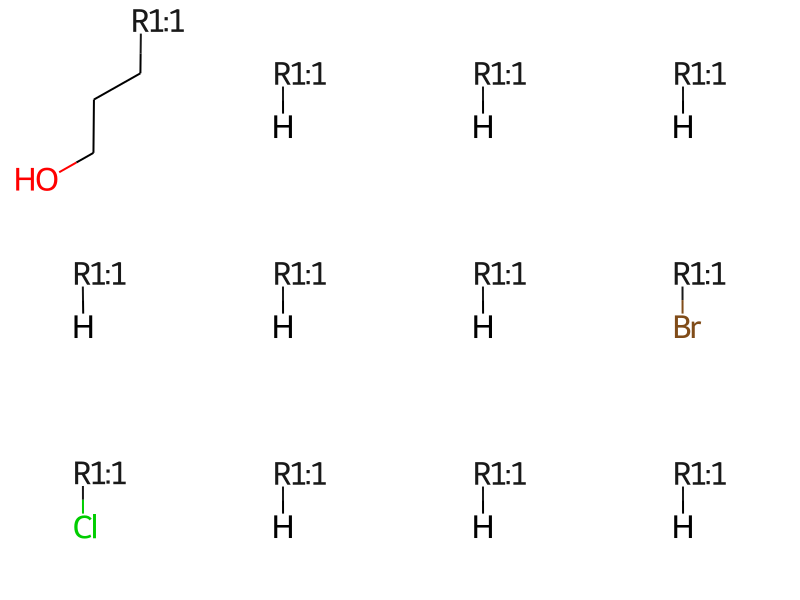
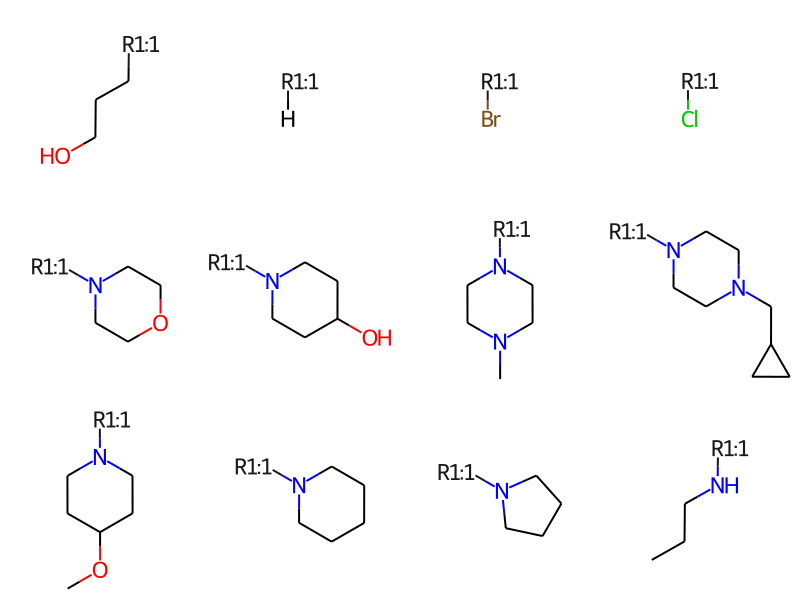
print(f'There are {len(rgd["R1"])} R1s')Draw.MolsToGridImage(rgd['R1'][:12],molsPerRow=4)
There are 25 R1s
Remove any R groups which have more than one dummy atom. This happens if an R group is attached to the core at multiple points and it may mess up the rest of the analysis.
qa = rdqueries.AtomNumEqualsQueryAtom(0)r1 = [x for x in rgd['R1'] iflen(x.GetAtomsMatchingQuery(qa))==1]r2 = [x for x in rgd['R2'] iflen(x.GetAtomsMatchingQuery(qa))==1]r3 = [x for x in rgd['R3'] iflen(x.GetAtomsMatchingQuery(qa))==1]
Now find the unique members in each set of R groups:
def uniquify(rgs): seen =set() keep = []for rg in rgs: smi = Chem.MolToSmiles(rg)if smi notin seen: keep.append(rg) seen.add(smi)return keep
Enumerating all possible molecules from the R groups
Quick intro to molzip
We’ll use the RDKit’s molzip() function to recombine the cores with the side chains.
molzip lets you take a molecule containing multiple fragments and “zip” them together. The atoms which should be bonded in the final molecule are labelled by connecting them to dummy atoms. The code identifies matching dummy atoms (by default this means dummies with the same isotopic label) in the fragments, adds bonds between the atoms connected to the dummies, and then removes the dummies.
Here’s a simple example using a molecule with three fragments:
And this is what happens when we zip those together:
Chem.molzip(sample)
Using molzip with RGD output
The molzip function is perfect for working with the output from an R-group decomposition.
Here I’ll define the function we’re going to use to enumerate all of the products:
import randomdef enumerate_all_products(core,*rgroups,randomOrder=False):# preserve the positions of the non-dummy core atoms, # we will use these to make sure the cores are drawn# the same way in each molecule we generate corePos = {} conf = core.GetConformer()for i inrange(conf.GetNumAtoms()): corePos[i] = Geometry.Point2D(conf.GetAtomPosition(i))# Python's itertools handles doing the combinatorics of generating# every possible combination of R groups: order = itertools.product(*rgroups)if randomOrder: order =list(order) random.shuffle(order)# now we just loop over each combination, copy all the pieces into# one molecule, and zip it. That's our productfor tpl in order: tm = Chem.RWMol(core)for r in tpl: tm.InsertMol(r) prod = Chem.molzip(tm)if prod isnotNone:# generate 2d coordinates with the core fixed in place rdDepictor.Compute2DCoords(prod,canonOrient=False,coordMap=corePos)# and finally yield the product moleculeyield prod
We’re going to use the core which came from the RGD since it’s labelled in the same way as the sidechains
rgd_core = rgd['Core'][0]rgd_core
scaffold
1
Let’s try it out:
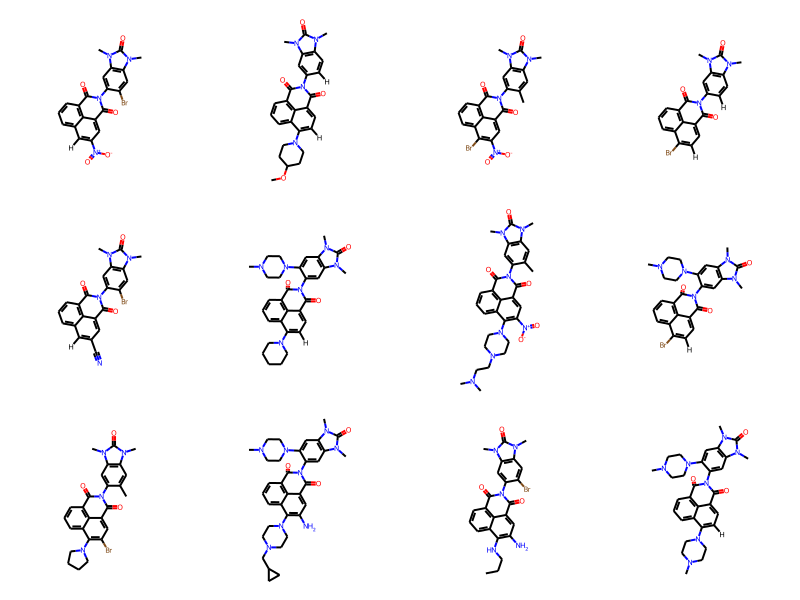
# this returns a generator which produces products:prod_gen = enumerate_all_products(rgd_core,r1s,r2s,r3s)# now get the first unique products prods = []seen =set()for prod in prod_gen:if prod isnotNone: Chem.SanitizeMol(prod) smi = Chem.MolToSmiles(prod)if smi notin seen: prods.append(prod) seen.add(smi)iflen(prods)>=12:breakDraw.MolsToGridImage(prods,molsPerRow=4)
Those come back ordered by the R groups (i.e. all products created using the first value of R1, then all products created using the second value of R1, etc.). This is fine if we’re planning on enumerating all the molecules, but if we only need a subset we can tell enumerate_all_products() to return the results in a random order:
# set a random seed so that we get reproducible resultsrandom.seed(0xbaff7ed)prod_gen = enumerate_all_products(rgd_core,r1s,r2s,r3s,randomOrder=True)prods = []seen =set()for prod in prod_gen:if prod isnotNone: Chem.SanitizeMol(prod) smi = Chem.MolToSmiles(prod)if smi notin seen: prods.append(prod) seen.add(smi)iflen(prods)>=12:breakDraw.MolsToGridImage(prods,molsPerRow=4)